









Bệnh Citrullinemia
15:50:3508/09/2014Bệnh thường biểu hiện trong vài ngày tuổi. Khi mới sinh, trẻ mắc bệnh trông khỏe mạnh bình thường nhưng amoniac tích tụ trong cơ thể gây ra: Thiếu năng lượng (thờ ơ), khó cho bú, nôn, co giật và mất ý thức. Trong nhiều trường hợp, bệnh trở nên rất nguy kịch
Giới thiệu bệnh CIT
Citrullinemia là một rối loạn di truyền gây tích tụ amoniac và những chất độc khác trong cơ thể. Nguyên nhân chính gây bệnh là do thiếu enzym argininosuccinate synthase 1 (AS), làm tích tụ citrulline trong máu và không đào thải được amoniac ra khỏi cơ thể. Bệnh gây tổn thương não và có thể tử vong nếu không được điều trị kịp thời.
Các dạng bệnh CIT và biểu hiện
Bệnh Citrullinemia có 2 dạng: Citrullinemia type I và Citrullinemia type II với các dấu hiệu và triệu chứng khác nhau gây ra bởi những đột biến khác nhau.
Citrullinemia type I (còn gọi là citrullinemia cổ điển) do đột biến trên gen ASS1 (nhiễm sắc thể 9) gây ra. Gen này mã hóa sản xuất enzym argininosuccinate synthase 1 giữ vai trò trong 1 bước của chu trình ure (citrulline chuyển thành axit argininosuccinic, có sự tham gia của axit aspartate). Đột biến trên gen ASS1 làm giảm hoạt lực của enzym, phá vỡ chu trình ure và ngăn cản cơ thể sản xuất nitơ. Nitơ dư thừa (dưới dạng amoniac) và các sản phẩm phụ của chu trình ure tích tụ trong máu.
Bệnh thường biểu hiện trong vài ngày tuổi. Khi mới sinh, trẻ mắc bệnh trông khỏe mạnh bình thường nhưng amoniac tích tụ trong cơ thể gây ra: Thiếu năng lượng (thờ ơ), khó cho bú, nôn, co giật và mất ý thức. Trong nhiều trường hợp, bệnh trở nên rất nguy kịch.
Một dạng ít gặp hơn và nhẹ hơn loại I có thể khởi phát muộn ở tuổi vị thành niên hoặc tuổi trưởng thành. Dạng khởi phát muộn này liên quan đến chứng đau đầu nghiêm trọng, thị giác kém, các vấn đề về cân bằng và liên kết cơ (mất điều hòa) và thờ ơ. Một số người mang đột biến gen gây bệnh citrullinemia type I không bao giờ biểu hiện bệnh.
Citrullinemia type II do đột biến trên gen SLC25A13 (nhiễm sắc thể 7) gây ra. Gen này mã hóa sản xuất protein citrin. Trong tế bào, citrin giúp vận chuyển glutamate vào trong ty thể và axit aspartate ra ngoài ty thể. Citrin còn được sử dụng trong việc sản xuất và phá vỡ cấu trúc đường đơn, hình thành các protein, nucleotide và tham gia chu trình ure. Do đột biến gen nên cơ thể thiếu citrin, dẫn đến ức chế chu trình ure và gây rối loạn hình thành protein và nucleotide.
Bệnh chủ yếu ảnh hưởng đến hệ thần kinh, gây khó ngủ, mất trí, cư xử bất thường (như hay gây gổ, dễ cáu bẳn, hiếu động thái quá), co giật và hôn mê. Trong một số trường hợp, các dấu hiệu và triệu chứng bệnh xuất hiện ở tuổi trưởng thành (khởi phát muộn). Các triệu chứng này có thể nguy kịch và được biết là gây ra bởi khi sử dụng một số loại thuốc, bị nhiễm trùng, phẫu thuật và uống rượu ở người khởi phát bệnh citrullinemia type II muộn.
Tỷ lệ mắc bệnh CIT
Citrullinemia type I có tỷ lệ mắc 1/57.000 trẻ được sinh ra.
Citrullinemia type II có tỷ lệ mắc 1/230.000 đến 1/100.000 trẻ.
Đặc điểm di truyền bệnh CIT
Các đột biến gen ASS1 và SLC25A13 gây ra bệnh CIT. Bệnh CIT là bệnh thuộc nhóm rối loạn chu trình chuyển hóa Ure do di truyền. Chu trình Ure xảy ra trong tế bào gan sử dụng những Nito thừa trong quá trình phân giải protein để tạo ra ure và bài tiết ra ngoài cơ thể cùng với nước tiểu. Các đột biến gen ASS1 làm giảm hoặc không tổng hợp enzyme argininosuccinate synthase tham gia 1 bước trong chu trình chuyển hóa Nito thành ammoniac dẫn đến tích tụ ammoniac và các sản phẩm thừa khá trong máu. Việc tích tụ này gây ra các triệu chứng thần kinh như mô tả ở loại I CIT.
Gen SLC25A13 mã hóa cho protein Citrin giúp các tế bào vận chuyển các chất được dùng trong chu trình Ure, hay vận chuyển nucleotide xây dựng nên ADN. Các đột biến SLS25A13 làm cho Citrin không được tổng hợp hoặc thiếu dẫn đến ức chế chu trình Ure, quá trình tạo ra protein hay vận chuyển nucleotide, tích tụ Amoniac và các biểu hiện của bệnh CIT lọa II.
Đặc điểm di truyền của bệnh CIT
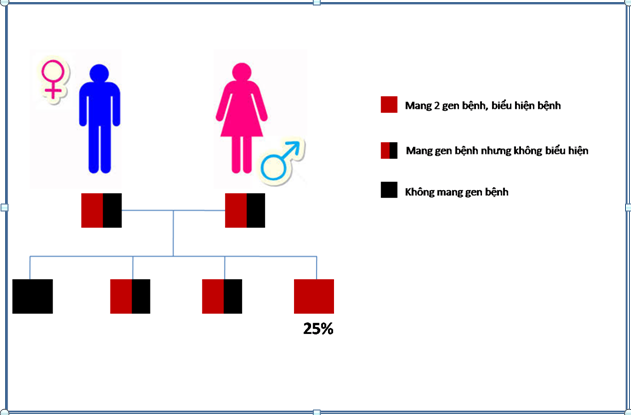
Bệnh CIT là bệnh di truyền tính trạng lặn trên nhiễm sắc thể thường, có nghĩa là để biểu hiện bệnh thì bệnh nhân phải có 2 bản sao của gen bệnh (hay đột biến) trên cặp nhiễm sắc thể tương đồng .Những người chỉ có 1 bản sao sẽ không biểu hiện bệnh nhưng có thể truyền cho con cái họ gẹn bệnh này. Khi cả bố và mẹ mang gen bệnh nhưng không biểu hiện thì tỷ lệ con của họ mắc bệnh là 25%.
Điều trị
Phát hiện sớm bằng gói sàng lọc sơ sinh 48 bệnh tại Trung Tâm Sàng Lọc Sơ Sinh Bionet Việt Nam Điều trị bằng cách hạn chế sử dụng một số protein trong thực phẩm và bổ sung arginine cùng một số thực phẩm thuốc để giảm tích tụ những tiền chất độc hại. Việc sử dụng các thực phẩm phải đảm bảo cân bằng để cung cấp đủ calo cho cơ thể.
- TỔNG QUAN XÉT NGHIỆM CHẨN ĐOÁN GENE BỆNH THIẾU MEN G6PD TẠI BIONET VIỆT NAM (13/05/2022)
- XÉT NGHIỆM CHẨN ĐOÁN SINH HÓA BỆNH THIẾU MEN G6PD – CẬP NHẬT NĂM 2022 (30/03/2022)
- NHỮNG LƯU Ý VỀ CHẾ ĐỘ DINH DƯỠNG, DÙNG THUỐC CỦA MẸ TRONG GIAI ĐOẠN CHO CON MẮC BỆNH THIẾU MEN G6PD BÚ MẸ (12/02/2022)
- LƯU Ý KHI SỬ DỤNG THUỐC TRỊ HO CHO NGƯỜI THIẾU MEN G6PD - NĂM 2022 (21/01/2022)
- NHỮNG LƯU Ý KHI SỬ DỤNG THUỐC GIẢM ĐAU/HẠ SỐT CHO NGƯỜI THIẾU MEN G6PD – CẬP NHẬT NĂM 2022 (20/01/2022)
Lượng truy cập hiện tại: 1
Lượng truy cập trong ngày: 4077
Tổng lượng truy cập: 14496304


