









BỆNH RỐI LOẠN CHU TRÌNH CHUYỂN HÓA URÊ BẨM SINH
17:09:1025/04/2016Nếu rối loạn chu trình chuyển hóa Urê không được phát hiện kịp thời sẽ để lại nhiều di chứng nặng nề cho trẻ và việc điều trị tốn kém. Vì vậy khi trẻ có dấu của bệnh cần đưa đến các bệnh viện chuyên khoa để được chẩn đoán đúng và điều trị kịp thời, bệnh nhi có thể được cứu sống và phát triển bình thường.
I. GIỚI THIỆU
Chu trình chuyển hóa Urê (UCDs) được mô tả đầu tiên vào năm 1932 bởi Krebs và Henseleit. Vai trò chính của chu trình Urê là chuyển hóa nitơ - một sản phẩm thải từ sự trao đổi chất protein và từ khẩu phần ăn thành urê. Ở người bình thường urê được bài tiết qua nước tiểu. Khiếm khuyết trong chu trình chuyển hóa Urê khiến nitơ không được chuyển hóa, bị tích tụ lại dưới dạng ammoniac là một chất độc đối với cơ thể, đặc biệt là đối với hệ thần kinh.
Đây là nhóm bệnh hiếm gặp, tỷ lệ mắc bệnh ước tính khoảng 1/25.000 – 1/30.000 trẻ sơ sinh. Các bệnh rối loạn chu trình chuyển hóa Urê nếu không phát hiện sớm và điều trị kịp thời có thể dẫn đến các biến chứng nghiêm trọng, thậm chí tử vong. Bệnh thường được điều trị bằng các phương pháp như: giảm nồng độ Nitơ trong huyết thanh, giảm lượng tiêu thụ các thực phẩm có chứa Nitơ trong khẩu phần ăn,…. Hiện nay cũng chưa có phương pháp chữa trị bệnh triệt để.
II. NGUYÊN NHÂN GÂY BỆNH
Khi vào cơ thể, protein được phân giải thành các axit amin và các axit amin tiếp tục được phân giải để cung cấp năng lượng cho các hoạt động cần thiết. Trong quá trình này nitơ (nitrogen) được sản sinh ra như một sản phẩm thải. Một chu trình chuyển hóa gồm nhiều phản ứng khác nhau gọi là chu trình chuyển hóa Urê có vai trò loại bỏ nitơ ra khỏi máu, chuyển hóa nitơ thành urê và được bài tiết qua thận dưới dạng nước tiểu.
Trong chu trình chuyển hóa Urê có sự tham gia của nhiều enzyme và cofactor đóng vai trò xúc tác cho các phản ứng. Ở người mắc bệnh, một trong số các enzyme tham gia vào chu trình bị mất hoặc giảm hoạt tính khiến cho các phản ứng trong chu trình không được diễn ra trọn vẹn dẫn đến việc tích tụ ammoniac (NH4). Ammoniac là một chất độc cơ thể không đào thải được. Ammoniac tích tụ ở nồng độ cao trong huyết tương sẽ gây đầu độc hệ thần kinh, ảnh hưởng đến nhiều bộ phận trong cơ thể.
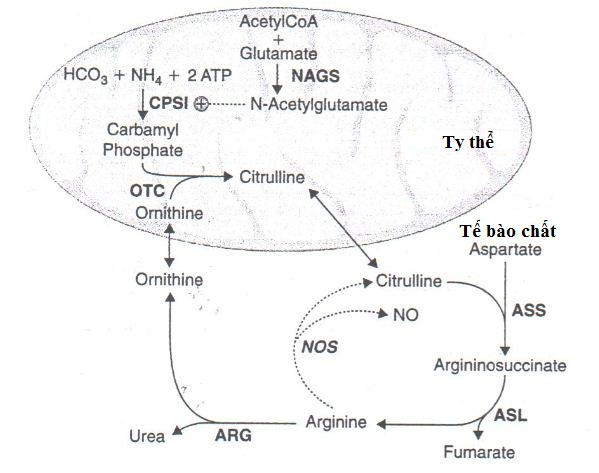
Hình minh họa: Các bước chính trong chu trình chuyển hóa Urê
Mức độ nặng hay nhẹ của bệnh phụ thuộc vào loại enzyme bị thiếu hụt và mức độ enzyme giảm hoạt tính. Dạng bệnh rối loạn nghiêm trọng xảy ra khi một trong bốn enzyme đầu tiên trong chu trình bị thiếu hụt nghiêm trọng hoặc mất toàn bộ hoạt tính bao gồm: enzyme carbamyl phosphate synthetase I (CPSI), orinithinetranscarbamylase (OTC), argininosuccinic acid synthetase (ASS), argininosuccinic acid lyase (ASL). Ở những dạng bệnh nhẹ hơn hoặc thiếu hụt enzyme một phần, các triệu chứng của bệnh có thể được kích hoạt bởi bệnh tật hoặc stress trong cuộc sống hằng ngày dẫn đến việc tăng nồng độ ammoniac trong huyết tương.
| PHÂN LOẠI BỆNH RỐI LOẠN CHUYỂN HÓA CHU TRÌNH URÊ | TỶ LỆ MẮC BÊNH | GENE | LOCUS |
| Thiếu hụt enzyme carbamyl phosphate synthetase I (CPSI) | 1/50.000 | CPSI | 2q35 |
| Thiếu hụt enzyme Orinithinetranscarbamylase (OTC) | 1/30.000 | OTC | Xp21.1 |
| Thiếu hụt enzyme Arigininosuccinatesynthetase hay chứng tăngcitrullinemia loại I (ASS hoặc CTLNI) | 1/50.000 | ASS | 9q34.1 |
| Thiếu hụt enzyme Argininosuccinate lyase (ASA) | 1/50.000 | ASL | 7cen-q11.2 |
| Thiếu hụt enzyme Arginase (ARGI) | 1/100.000 | ARGI | 6q23 |
| Thiếu hụt enzyme N-Acetyl glutamate synthase (NAGS) | 1/100.000 | NAGS | 17q21.31 |
| Hội chứng Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) | 1/100.000 | SLC25A15 | 13q.14 |
| Chứng tăng Citrullinemia loại II (CTLNII) | 1/100.000 | SLC25A13 | 7q21.3 |
Bảng phân loại bệnh thuộc nhóm bệnh rối loạn chuyển hóa chu trình Urê và các gene liên quan
Cơ sở phân tử của bệnh là do đột biến trong gene quy định việc tổng hợp các enzyme, cofactor tham gia vào chu trình chuyển hóa Urê. Hầu hết các bệnh trong nhóm bệnh UCDs là bệnh di truyền tính trạng lặn trên nhiễm sắc thể (NST) thường, ngoại trừ bệnh OTC là bệnh di truyền liên kết với NST giới tính X. Với bệnh di truyền tính trạng lặn trên NST thường để biểu hiện bệnh thì bệnh nhân phải nhận 2 bản sao của gene bệnh (hay đột biến) từ cả bố và mẹ. Những người chỉ có 1 bản sao của gene bệnh sẽ không biểu hiện bệnh và được gọi là thể mang, những người này có thể truyền gene bệnh cho con cái họ. Khi cả bố và mẹ là thể mang thì tỷ lệ con của họ mắc bệnh là 25%.
III. TRIỆU CHỨNG BỆNH
Tùy thuộc vào loại enzyme bị thiếu hụt và mức độ thiếu hụt bệnh có thể biểu hiện triệu chứng ngay sau khi sinh hoặc ở giai đoạn muộn hơn trong thời niên thiếu hoặc trưởng thành.
Giai đoạn sơ sinh khởi phát:
Những dấu hiệu điển hình của bệnh trong giai đoạn sơ sinh thường là kết quả của những khiếm khuyết nghiêm trọng trong chu trình Urê. Việc nhận biết bệnh ở giai đoạn sơ sinh gặp nhiều khó khăn do các dấu hiệu của bệnh khá giống với các triệu chứng của các bệnh thông thường khác như: nhiễm trùng sơ sinh hay chứng giảm oxy sơ sinh. Những trẻ sơ sinh mắc bệnh UCDs thường có vẻ ngoài bình thường sau khi sinh nhưng dần dần sức khỏe sẽ xấu đi khi ammoniac bị tích tụ trong cơ thể. Những dấu hiệu đầu tiên thường diễn ra sau 48 đến 72 giờ sau sinh khi sự hấp thu dinh dưỡng của trẻ tăng lên nhưng cũng có thể trì hoãn đến khoảng 1 - 2 tuần mới biểu hiện. Khởi đầu bệnh nhân có biểu hiện khó chịu, trẻ bú kém, bỏ bú hoặc nôn mửa, ngủ mơ màng hoặc ngủ li bì. Ngay sau đó cơn co giật, giảm trương lực (yếu cơ bắp), suy hô hấp, hôn mê có thể xảy ra. Những dấu hiệu này là do sự tăng nồng độ ammoniac trong máu gây đầu độc hệ thần kinh và các cơ quan. Những bệnh nhân không được kiểm soát kịp thời sẽ rơi vào hôn mê và tử vong.
Hình minh họa : Nếu bệnh không được kiểm soát kịp thời trẻ có thể rơi vào hôn mê và tử vong.
Những triệu chứng ở giai đoạn sơ sinh thường gặp nhất ở trẻ nam mắc bệnh thiếu hụt enzyme OTC là bệnh di truyền liên kết với NST giới tính X.
Giai đoạn khởi phát muộn:
Bệnh nhân không có triệu chứng trong giai đoạn sơ sinh mà biểu hiện bệnh muộn hơn thường do các enzyme trong chu trình urê bị giảm hoạt tính ít, còn hoạt động được một phần. Bệnh chỉ khởi phát khi có tác động của một số yếu tố như sự suy giảm hoạt tính enzyme, chế độ ăn, các yếu tổ ảnh hưởng từ môi trường. Ngoài ra việc biểu hiện triệu chứng bệnh có thể được kích hoạt khi bệnh nhân sử dụng một số loại thuốc, hóa dược gây ảnh hưởng đến chức năng của chu trình Urê như thuốc valproic hoặc khi mang thai. Các biểu hiện sớm có thể bao gồm hành vi hiếu động, đôi khi kèm theo la hét và hành vi tự gây thương tích, từ chối ăn thịt hoặc các loại thực phẩm giàu protein khác. Những triệu chứng muộn sau đó bao gồm thường xuyên nôn mửa (đặc biệt là sau bữa ăn có hàm lượng protein cao), mê sảng và cuối cùng nếu không được chẩn đoán và điều trị kịp thời bệnh nhân rơi vào tình trạng hôn mê và tử vong.
IV. SÀNG LỌC VÀ CHẨN ĐOÁN BỆNH
1. Sàng lọc bệnh
Sàng lọc bệnh ở trẻ sơ sinh thường được thực hiện bằng việc sử dụng hệ thống khối phổ MS – MS để phát hiện sự có mặt các chất liên quan đến chu trình Urê có trong máu. Hiện nay sàng lọc sơ sinh được áp dụng cho bệnh thiếu hụt enzyme arginase (ARG), Argininosuccinate lyase (ASA), CPS, citrullinemia loại I (CIT-I), hội chứng HHH.
2. Chẩn đoán bệnh
Bước quan trọng nhất trong việc chẩn đoán bệnh là xác định các dấu hiệu lâm sàng của chứng tăng ammoniac máu. Xác định nồng độ ammoniac trong máu là xét nghiệm đầu tiên cần tiến hành với bệnh nhân nghi ngờ mắc bệnh rối loạn chu trình Urê.
Ngoài nồng độ ammoniac một số dữ liệu khác cũng phục vụ cho việc chẩn đoán bệnh UCDs như độ pH, nồng độ CO2, khoảng trống anion (anion gap), nồng độ axit lactic trong máu, acylcarnitine, nồng độ amino axit trong máu và nước tiểu, xét nghiệm axit hữu cơ trong nước tiểu đặc biệt là axit orotic.
Chẩn đoán gene và enzyme cũng được dùng nhằm xác định tất cả các bệnh trên. Với bệnh thiếu hụt CPSI, OTC và NAGS, phương pháp chẩn đoán enzyme được thực hiện trên mẫu sinh thiết gan giữ trong nitơ lỏng. Phương pháp phân tích enzyme trong nguyên bào sợi được thực hiện với bệnh ASS và ASL được thực hiện với nguyên bào sợi, với bệnh ARGI và ASL có thể thực hiện trên tế bào máu.
Xét nghiệm trước sinh với bệnh UDCs bao gồm việc xác định các chất tích tụ trong dịch ối như ASA trong bệnh ASL, phân tích enzyme trong tế bào lông nhung màng đệm, tế bào máu thai nhi và xét nghiệm ADN tự do của thai nhi có trong máu mẹ.
V. PHƯƠNG PHÁP ĐIỀU TRỊ
Các biện pháp cấp cứu khẩn cấp với bệnh nhân mắc UCDs tuân thủ theo 3 nguyên tắc chính sau:
- Các biện pháp loại bỏ ammoniac bằng lọc máu hoặc các phương pháp tương tự.
- Đảo ngược quá trình dị hóa bằng việc cung cấp nguồn năng lượng cao và trong một số trường hợp đặc biệt là ức chế hoạt động hormone (insulin).
- Các biện pháp dược học sử dụng các loại thuốc thải độc nitơ.
Các phương pháp điều trị bệnh UCDs dài hạn bao gồm chế độ ăn hạn chế protein (trong thức ăn), bổ sung các thuốc, thực phẩm chứa amino axit thiết yếu, thực phẩm cung cấp năng lượng, bổ sung vitamin và khoáng chất và sử dụng các thuốc thải độc nitơ, Việc duy trì khẩu phần ăn hạn chế protein tùy thuộc vào loại enzyme bị thiếu hụt trong chu trình Urê (bệnh nhân mắc bệnh OTC hoặc CPS cần hạn chế protein nhiều nhất) và cần phù hợp với thể trạng bệnh nhân (chiều cao, cân nặng, chu vi vòng đầu, nồng độ amino axit và ammoniac trong máu, tổng lượng protein và albumin , tỷ lệ thể tích hồng cầu, hàm lượng sắt và prealbumin).

Chế ăn hạn chế protein là điều cần thiết với người mắc bệnh UCDs
Ngoài ra trẻ mắc bệnh cũng cần được kiểm tra định kỳ để đảm bảo lượng protein cơ thể hấp thu trong khoảng hợp lý và quá trình trao đổi chất được kiểm soát tốt. Các đánh giá lâm sàng cần tiến hành bao gồm: mức tăng cân nặng, tăng chiều cao, chu vi vòng đầu (đối với trẻ sơ sinh), kích thước gan, vẻ ngoài của tóc, da và móng. Các đánh giá bệnh học thần kinh nên bắt đầu vào khoảng 1 năm tuổi. Ngoài ra cũng cần kiểm tra các chỉ số sinh hóa như nồng độ ammoniac trong huyết tương, định lượng amino axit, men gan và tổng hàm lượng protein, albumin, prealbumin và sắt.
Lời kết: Rối loạn chu trình chuyển hóa Urê bẩm sinh là nhóm bệnh nguy hiểm tuy nhiên triệu chứng bệnh không điển hình nên dễ bị bỏ sót và nhầm lẫn với các bệnh lý khác. Trước những tổn thương thần kinh trung ương không giải thích được ở trẻ như biểu hiện bú kém, bỏ bú, li bì, hôn mê, vàng da, đái tháo đường, nhiễm trùng huyết, trẻ thở nhanh hoặc ngừng thở (giống biến chứng của ngạt nặng sau sinh) mặc dù trẻ không có tiền sử bị ngạt khi sinh, rối loạn nhịp tim… mà qua khám không xác định được nguyên nhân thì phải nghĩ tới bệnh rối loạn chuyển hóa bẩm sinh và đưa trẻ đi làm xét nghiệm chuyên sâu để được chẩn đoán bệnh.
Nếu rối loạn chu trình chuyển hóa Urê không được phát hiện kịp thời sẽ để lại nhiều di chứng nặng nề cho trẻ và việc điều trị tốn kém. Vì vậy khi trẻ có dấu của bệnh cần đưa đến các bệnh viện chuyên khoa để được chẩn đoán đúng và điều trị kịp thời, bệnh nhi có thể được cứu sống và phát triển bình thường.
Trung tâm SLSS Bionet Việt Nam hiện đang cung cấp gói xét nghiệm cao cấp 48 bệnh phát hiện được hầu hết các bệnh liên quan đến chu trình chuyển hóa Urê. Chi tiết xem tại đây.
Nguồn: Bionet
Tài liệu tham khảo
1. http://nutrition.nutricia.com/conditions/urea-cycle-disorders#causes
2. https://en.wikipedia.org/wiki/Urea_cycle_disorder
3. “Pediatric Endocrinology and Inborn Errors of Metabolism” - Kyriakie Sarafoglou, Georg Hoffmann, Karl Roth
- TỔNG QUAN XÉT NGHIỆM CHẨN ĐOÁN GENE BỆNH THIẾU MEN G6PD TẠI BIONET VIỆT NAM (13/05/2022)
- XÉT NGHIỆM CHẨN ĐOÁN SINH HÓA BỆNH THIẾU MEN G6PD – CẬP NHẬT NĂM 2022 (30/03/2022)
- NHỮNG LƯU Ý VỀ CHẾ ĐỘ DINH DƯỠNG, DÙNG THUỐC CỦA MẸ TRONG GIAI ĐOẠN CHO CON MẮC BỆNH THIẾU MEN G6PD BÚ MẸ (12/02/2022)
- LƯU Ý KHI SỬ DỤNG THUỐC TRỊ HO CHO NGƯỜI THIẾU MEN G6PD - NĂM 2022 (21/01/2022)
- NHỮNG LƯU Ý KHI SỬ DỤNG THUỐC GIẢM ĐAU/HẠ SỐT CHO NGƯỜI THIẾU MEN G6PD – CẬP NHẬT NĂM 2022 (20/01/2022)
Lượng truy cập hiện tại: 1
Lượng truy cập trong ngày: 1827
Tổng lượng truy cập: 15252052


